RNA-seq Tutorial- STAR, StringTie and DESeq2*
New RNA-seq tuxedo protocol using the Discovery Environment
Rational and background
RNA-seq involves preparing the mRNA which is converted to cDNA and provided as input to next generation sequencing library preparation method. Prior to RNA-seq there were hybridization based microarrays used for gene expression studies, the main drawback was the poor quantification of lowly and highly expressed genes. RNA-seq provides distinct advantages over microarrays, it provides better insights into alternative gene splicing, post-transcriptional modifications, gene fusion and deferentially expressed genes and thus helping to understanding the gene structure and expression patterns of genes across different samples, treatment conditions and time points. The ease of sequencing and the low cost have made RNA-seq a workhorse in transcriptomic studies and viable option even for small scale labs. But the main challenge remains in analyzing the sequenced data.
The current ecosystems of RNA-seq tools provide a varied ways of analyzing RNA-seq data. Depending on the experiment goal one could align the reads to reference genome or pseduoalign to transcriptome and perform quantification and differential expression of genes or if you want to annotate your reference, assemble RNA-seq reads using a denvo transcriptome assembler. Here we focus on workflows that align reads to reference genomes. The most commonly cited and widely used workflow is the Tuxedo protocol (Tophat,Cufflinks,Cuffdiff)developed by Cole Trapnell et al. The main drawback of this workflow is the ability to scale i.e they tend to take more runtime compared to newly updated Tuxedo protocol(HISAT,StringTie,Ballgown) by Mihaela Pertea et al. This updated Tuxedo protocol not only scales but is more accurate in detecting deferentially expressed genes.Another alternate workflow uses STAR as the aligner instead of HISAT2 in the updated Tuxedo workflow.
In this example we will compare gene transcript abundance drought sensitive sorghum line under drought stress(DS) and well-watered (WW) condition. The expression of drought-related genes was more abundant in the drought sensitive genotype under DS condition compared to WW.
We will use RNAseq to compare expression levels for genes between DS and WW- samples for drought sensitive genotype IS20351 and to identify new transcripts or isoforms. In this tutorial, we will use data stored at the NCBI Sequence Read Archive.
- Align the data to the Sorghum v1 reference genome using STAR
- Transcript assembly using StringTie
- Identify differential-expressed genes using Ballgown
- Use Atmosphere to visually explore the differential gene expression results.
If you do not have an account, please see one of the on-site CyVerse staff for a temporary account.
Specific Objectives
By the end of this module, you should
- Be more familiar with the DE user interface
- Understand the starting data for RNA-seq analysis
- Be able to align short sequence reads with a reference genome in the DE
- Be able to analyze differential gene expression in the DE and Atmosphere
Note on Staged Data:
Several of the methods in this tutorial can take 2 to 4 hrs to complete on a full-sized data set. So that you can complete the tutorial in the allotted time, we have pre-staged input and output files in the 'Community Data' folder for each step. You can start your analyses then skip to the next step using pre-staged data.
Original data from NCBI Sequence Read Archive study re accessible through GEO Series accession number GSE80699
- GSM2133750 IS20351_WW_1
- GSM2133751 IS20351_WW_2
- GSM2133752 IS20351_WW_3
- GSM2133753 IS20351_DS_1
- GSM2133754 IS20351_DS_2
- GSM2133755 IS20351_DS_3
Paper Reference for dataset: Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE,
Alessandra Fracasso, Luisa M. Trindade and Stefano Amaducci; DOI: 10.1186/s12870-016-0800-x, May 2016
The Staged Fastq Data can be found in the
Community Data -> iplantcollaborative -> example_data -> STAR-StringTie-DESeq2 -> reads

Section 1: Align reads to reference using STAR aligner
Spliced Transcripts Alignment to a Reference (STAR) software is another highly cited splice-ware aligner. It scores above the other aligners in terms of its speed of alignment. Its algorithm uses sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. STAR mapping workflow involves two steps i.e generating genome index files and then mapping the reads against the genome. STAR be found by navigating to Apps window by clicking on the icon
STAR paper: STAR: ultrafast universal RNA-seq aligner
Reference Manual
a) Open STAR app
Click on Operation -> Categories -> Mapping -> STAR-2.5.3a-index-align.

b) Click on the name of the App to open it.
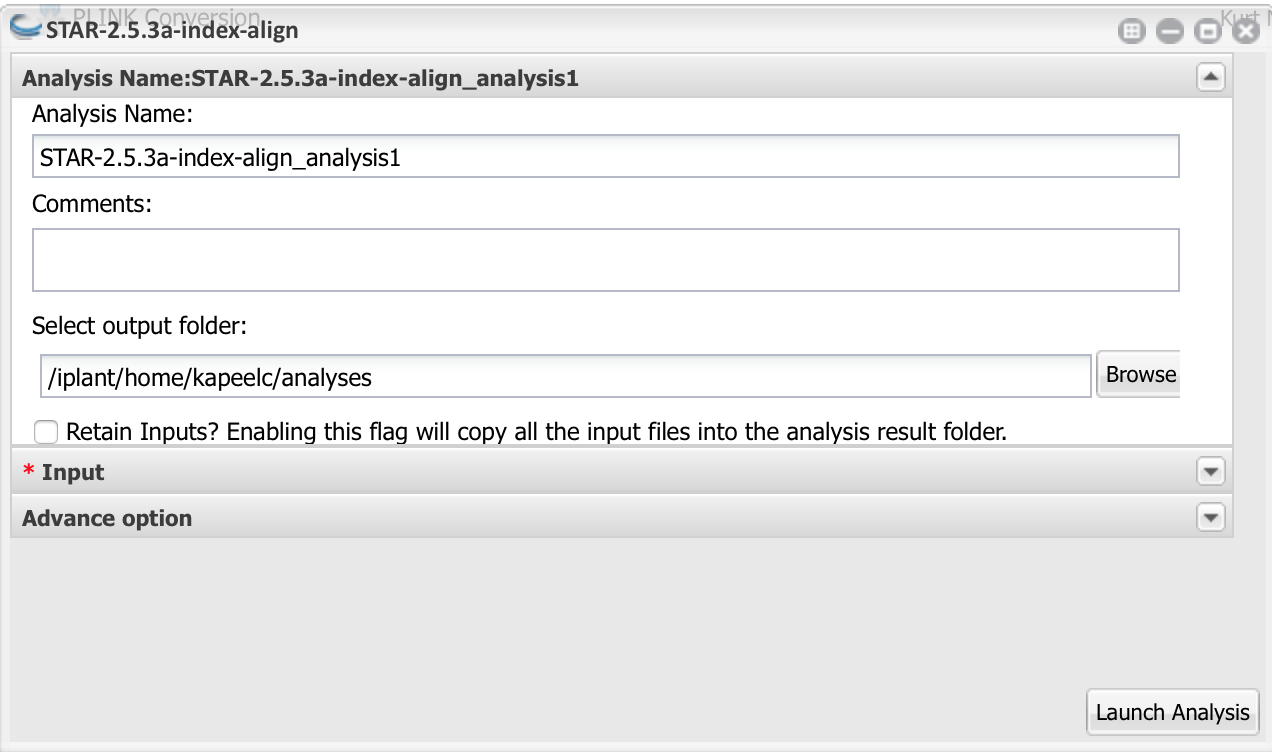
c) In the Input section,you need to first select the a reference genome and annotation file either from the drop list or upload a fasta for the genomes or gtf annotation file.

Here we upload the two files from:
Reference genome : Community Data ->iplantcollaborative->example_data->STAR-StringTie-DESeq2->Sorghum_bicolor.Sorbi1.20.dna.toplevel.fa Reference annotation : Community Data ->iplantcollaborative->example_data->STAR-StringTie-DESeq2->Sorghum_bicolor.Sorbi1.20.gtf

you can click 'Add' at the top right of the FASTQ File(s) box to navigate to the folder containing the FASTQ files: For paired end data add the read 1 gzipped files for all samples then followed by read 2 files in gzipped format.
Select File type as PE since we are using paired end data set.

Note: For multiple input files, it is convenient to open a separate data window, navigate to the above folder and drag all of files into the input data box. Each of the input files will be processed independently. For convenience, a batch of FASTQ files can be analyzed together but these files can also be processed concurrently in independent STAR runs
d) Start the analysis by clicking the Launch Analysis button, naming the analysis 'STAR' in the dialog box. Reasonable default options are provided for the analysis settings.

e) STAR will require some time to complete its work on each sample. Click on the Analyses icon to view the status of your submitted analysis.

f) When the analysis is complete, navigate to the results under your 'analyses' folder. The principle outputs from STAR are BAM files, each set of alignments is in its own folder, corresponding to the name of the original Fastq file. This is the most time-consuming step of this training module. If your analysis has not completed in time, you can skip ahead by using the pre-computed results:
Community Data ->iplantcollaborative->example_data->STAR-StringTie-DESeq2-> STAR_results
In the STAR_results folder, you should see these folders:

STAR_results: The result directory for the STAR runs contain the following-
index: STAR genome indices
bam_output: directory of alignment files coordinate sorted in bam format for each sample
output: Default output files from STAR which includes for each sample
Log.out: main log file with a lot of detailed information about the run. This file is most useful for troubleshooting and debugging.
Log.progress.out: reports job progress statistics, such as the number of processed reads, % of mapped reads etc.
Log.final.out: summary mapping statistics after mapping job is complete, very useful for quality control.
Aligned.out.sam - alignments in standard SAM format.
SJ.out.tab- only those reads that contain junctions.
ReadsPerGene.out.tab-read counts per gene
We will use the bam_output folder to assemble transcripts using Stringtie.
Section 2: Assemble transcripts with StringTie-1.3.3
StringTie like Cufflinks2 assembles transcripts from RNA seq reads aligned to the reference and does quantification. It follows a netflow algorithm where it assembles and quantitates simultaneously the highly expressed transcripts and removes reads associated with that transcripts and repeats the process until all the reads are used. This algorithm improves the run time for StringTie using less memory compared to Cufflinks2. If provided with a reference annotation file Stringtie uses it to construct assembly for low abundance genes, but this is optional. Alternatively, you can skip the assembly of novel genes and transcripts, and use StringTie simply to quantify all the transcripts provided in an annotation file
a) Click on the Apps icon and find StringTie-1.3.3. Open it. Name the analysis as StringTie-1.3.3

b) In the 'Select Input data' section, add the 'bam' files by navigating to the bam_output folder from STAR (above) and select and drag all bam files into the input box. For convenience, a batch of STAR bam files can be analyzed together but these files can also be processed concurrently in independent StringTie runs. The path for the STAR bam files
Community Data ->iplantcollaborative->example_data->STAR-StringTie-DESeq2-> STAR_results -> bam_output

c) In the Reference Annotaiton section, select reference annotation Sorghum_bicolor.Sorbi1.20.gtf.

d) Run StringTie-1.3.3. When it is complete, you will see the following outputs

StringTie_output contains the following files:
- Stringtie's main output is a GTF file containing the assembled transcripts e.g:IS20351_DS_1_1.gtf
- Gene abundances in tab-delimited format e.g IS20351_DS_1_1_abund.tab
- Fully covered transcripts that match the reference annotation, in GTF format e.g IS20351_DS_1_1.refs.gtf
Examine the GTF file, IS20351_DS_1_1.gtf This file contains annotated transcripts assembled by StringTie-1.3.3, using the annotated transcripts selected from the reference file uploaded in the StringTie-1.3.3 app as a guide.This file gives normalized expression metrics in both FPKM and TPM along with per base coverage
# StringTie version 1.3.3 8 StringTie transcript 10053 10774 1000 - . gene_id "STRG.1"; transcript_id "STRG.1.1"; cov "2.702216"; FPKM "0.811937"; TPM "0.509185"; 8 StringTie exon 10053 10774 1000 - . gene_id "STRG.1"; transcript_id "STRG.1.1"; exon_number "1"; cov "2.702216"; 8 StringTie transcript 11084 11299 1000 . . gene_id "STRG.2"; transcript_id "STRG.2.1"; cov "3.685185"; FPKM "1.107291"; TPM "0.694408"; 8 StringTie exon 11084 11299 1000 . . gene_id "STRG.2"; transcript_id "STRG.2.1"; exon_number "1"; cov "3.685185"; 8 StringTie transcript 13293 13892 1000 - . gene_id "STRG.3"; transcript_id "STRG.3.1"; cov "7.450417"; FPKM "2.238633"; TPM "1.403900"; 8 StringTie exon 13293 13892 1000 - . gene_id "STRG.3"; transcript_id "STRG.3.1"; exon_number "1"; cov "7.450417"; 8 StringTie transcript 38190 40022 1000 - . gene_id "STRG.4"; transcript_id "STRG.4.1"; reference_id "Sb08g000230.1"; ref_gene_id "Sb08g000230"; ref_gene_name "Sb08g000230"; cov "0.107474"; FPKM "0.032293"; TPM "0.020252"; 8 StringTie exon 38190 40022 1000 - . gene_id "STRG.4"; transcript_id "STRG.4.1"; exon_number "1"; reference_id "Sb08g000230.1"; ref_gene_id "Sb08g000230"; ref_gene_name "Sb08g000230"; cov "0.107474";
Section 3: Merge all StringTie-1.3.3 transcripts into a single transcriptome annotation file using StringTie-1.3.3_merge
StringTie-merge like Cufflinks-merge uses the same principle of merging the transcript assemblies from samples into a consolidated annotation set. This merging steps helps restore full length of the structure transcript especially for transcript assembled with low coverage.The main purpose of this application is to make it easier to make an assembly GTF file suitable for use with Ballgown. A merged, empirical annotation file will be more accurate than using the standard reference annotation, as the expression of rare or novel genes and alternative splicing isoforms seen in this experiment will be better reflected in the empirical transcriptome assemblies.
a) Open the StringTie-1.3.3_merge app. Under 'Input Data', browse to the results of the StringTie-1.3.3 analyses (gtf_out). Select the all GTF files as input for StringTie-1.3.3_merge.

b) Under Reference Annotation, select the Sorghum_bicolor.Sorbi1.20.gtf.
c) Run the App. When it is complete, you should see the following outputs under StringTie-1.3.3_merge_results:

We will use the merged.out.gtf with StrignTie-1.3.3 app again
Section 4: Create Ballgown input files using with StringTie-1.3.3
We will use StringTie-1.3.3 again to assemble transcripts but this time will will use the consolidated annotation file we got from StringTie-1.3.3_merge step. This will re-estimate the transcript abundances using the merged structures but reads may need to be re-allocated for transcripts whose structures were altered by the merging step. We will set the option -B, -e of StringTie which will create table count files (*ctab files) for each sample to be used in Ballgown for differential expression.

Run the App. When it is complete, you should see the following outputs under
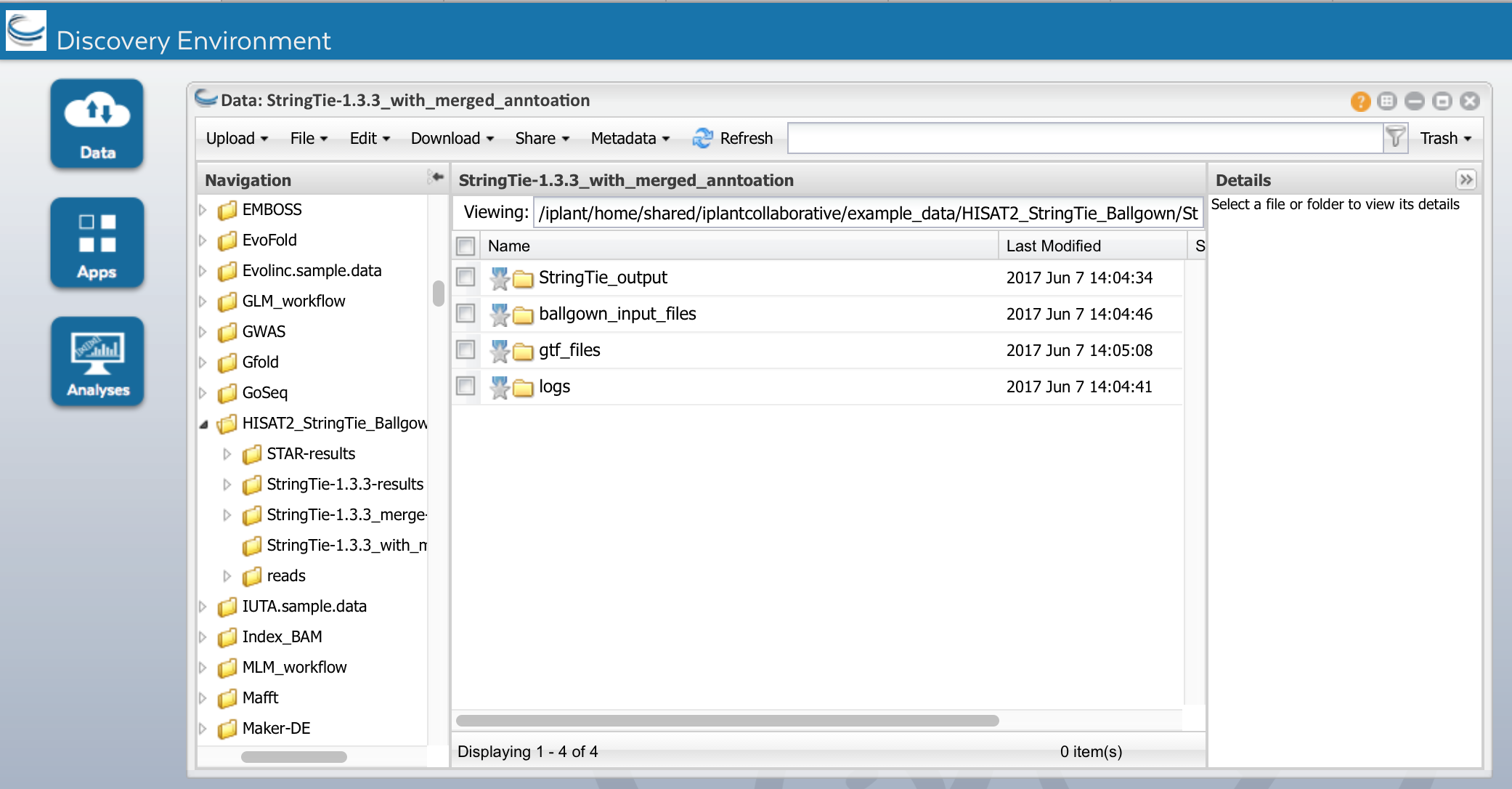
Ballgown_input_files: count files (*ctab files) for each sample
e_data.ctab: exon-level expression measurements.
i_data.ctab: intron- (i.e., junction-) level expression measurements
t_data.ctab: transcript-level expression measurements
e2t.ctab: table with two columns, e_id and t_id, denoting which exons belong to which transcripts
i2t.ctab: table with two columns, i_id and t_id, denoting which introns belong to which transcripts
More details of the ctab files please refere the Ballgown documentation
Section 5: Compare expression analysis using Ballgown
Ballgown is a R package that uses abundance data produced by StringTie to perform differential expression analysis at gene, transcript, exon or junction level. It does both time series and fixed condition differential expression analysis.
a) In Apps select Ballgown
b) provide a experiment design matrix file in txt, This file defines samples, group and replicate information e:g
ID condition reps
IS20351_DS_1_1 DS 1
IS20351_DS_1_2 DS 2
IS20351_DS_1_3 DS 3
IS20351_WW_1_1 WW 1
IS20351_WW_1_2 WW 2
IS20351_WW_1_3 WW 3
Here we are doing a pairwise comparison for differential expression in sensitive genotype under Drought Stress(DS) and Well Watered(WW) condition. We have 3 replicates under each condition.
c) Upload the design matrix file and provide experimental covariate which is condition(it should match column in design matrix file)

d) Launch analysis
e) Examine results

Successful execution of the Ballgown will create a directory named output. The directory will contain the following files:
- Rplots.pdf- Boxplot of FPKM distribution of each smaple
- results_gene.tsv- Gene level Differential expression with no filtering
- results_gene_filter.sig.tsv- Identify genes with p value < 0.05
- results_gene_filter.tsv- Filter low-abundance genes, here we remove all genes with a variance across samples less than one
- results_trans.tsv-transcript level Differential expression with no filtering
- results_trans_filter.sig.tsv- Identify transcripts with p value < 0.05
- results_trans_filter.tsv-Filter low-abundance genes, here we remove all transcript with a variance across samples less than one
RNA-Seq visualization in Atmosphere.
Section 1: Connect to an instance of an Atmosphere Image
1. Go to https://atmo.cyverse.org/ and log in with IPLANT TEST USER CREDENTIALS.

2. Click on images. Search "rnaseq" on the search space. Click on "RNAseq Differential Expression" image.

3. Click on Launch. Add to existing projects if you have already created one. In this case, we will add the instance to existing project "RNA-seq visualization". Select the size of the instance "tiny 2(CPU:1,Mem: 8, Disk: 60Gb)". Click on Launch instance. This will launch a instance of the image.
