RNA seq tutorials- Kallisto and Sleuth*
This tutorial is for using Kallisto and Sleuth RNA seq tool in DE
New RNA-seq tuxedo protocol using the Discovery Environment
Rational and background
RNA-seq involves preparing the mRNA which is converted to cDNA and provided as input to next generation sequencing library preparation method. Prior to RNA-seq there were hybridization based microarrays used for gene expression studies, the main drawback was the poor quantification of lowly and highly expressed genes. RNA-seq provides distinct advantages over microarrays, it provides better insights into alternative gene splicing, post-transcriptional modifications, gene fusion and deferentially expressed genes and thus helping to understanding the gene structure and expression patterns of genes across different samples, treatment conditions and time points. The ease of sequencing and the low cost have made RNA-seq a workhorse in transcriptomic studies and viable option even for small scale labs. But the main challenge remains in analyzing the sequenced data.
The current ecosystems of RNA-seq tools provide a varied ways of analyzing RNA-seq data. Depending on the experiment goal one could align the reads to reference genome or pseduoalign to transcriptome and perform quantification and differential expression of genes or if you want to annotate your reference, assemble RNA-seq reads using a denvo transcriptome assembler. Here we focus on workflows that pseudo align reads to reference transcriptome. The most commonly cited and widely used quantification workflow is Kallisto and Sleuth.
In this example we will compare gene transcript abundance drought sensitive sorghum line under drought stress(DS) and well-watered (WW) condition. The expression of drought-related genes was more abundant in the drought sensitive genotype under DS condition compared to WW.
We will use RNAseq to compare expression levels for genes between DS and WW- samples for drought sensitive genotype IS20351 and to identify new transcripts or isoforms. In this tutorial, we will use data stored at the NCBI Sequence Read Archive.
- build a transcriptome index using Kallisto index
- quantify abundances of transcripts using Kallisto qaunt
- Identify differential-expressed genes using Sleuth R package
- Use Atmosphere to visually explore the differential gene expression results.
If you do not have an account, please see one of the on-site CyVerse staff for a temporary account.
Specific Objectives
By the end of this module, you should
- Be more familiar with the DE user interface
- Understand the starting data for RNA-seq analysis
- Be able to pseduo align short sequence reads and quantify reference transcripts in the DE
- Be able to analyze differential gene expression in the DE and Atmosphere
Note on Staged Data:
Several of the methods in this tutorial can take 2 to 4 hrs to complete on a full-sized data set. So that you can complete the tutorial in the allotted time, we have pre-staged input and output files in the 'Community Data' folder for each step. You can start your analyses then skip to the next step using pre-staged data.
Original data from NCBI Sequence Read Archive study re accessible through GEO Series accession number GSE80699
- GSM2133750 IS20351_WW_1
- GSM2133751 IS20351_WW_2
- GSM2133752 IS20351_WW_3
- GSM2133753 IS20351_DS_1
- GSM2133754 IS20351_DS_2
- GSM2133755 IS20351_DS_3
Paper Reference for dataset: Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE,
Alessandra Fracasso, Luisa M. Trindade and Stefano Amaducci; DOI: 10.1186/s12870-016-0800-x, May 2016
The Staged Data can be found in the
Community Data -> iplantcollaborative -> example_data -> Kallisto_Sleuth
Kallisto and Sleuth RNA seq analysis
The kallisto-sleuth pipeline is quite simple. There are basically two steps:
- Kallisto indexing and qauntification (once per organism or annotation)
- Sleuth for analyzing gene level differential expression (once per each experimental sample)
You may have noticed that there is no alignment step. This is because part of the kallisto algorithm performs a very fast "alignment" which we call the pseudoalignment. This simplifies things from the user's point of view since there are no extra intermediate files.
1. Kallisto indexing and qauntification-
Before you can quantify with kallisto, you must create an index from an annotation file. For RNA-Seq, an annotation file is the set of cDNA transcripts in FASTA format (the "transcriptome"). If you are not working with a model organism, one option is to do a de novo assembly. Kallisto-index simply takes a FASTA file and outputs an index in a binary format that is designed for kallisto. There is no default extension for the index.
Once you index an annotation, you can quantify any number of samples against it. The quantification step includes pseudoalignment as well as running the EM algorithm to do estimation of transcript level abundances. THis particular app provides both indexing and qauntification in one step.The parameters are pretty minimal. You must supply an transcript file, an output location, and a set of reads. In the case of single-end data, you must use the option single under input , as well provide a fragment length distribution of reads. In the case of paired-end data use option paired under input, and the tool can infer the fragment length distribution from the data.
There is also one other important parameter: the number of bootstrap iterations. By default, kallisto runs zero bootstrap iterations. If you do not plan to run sleuth for differential expression analysis, this is okay. But if you plan to run sleuth, you must provide a nonzero number of bootstraps. In general, this number should be at least 30. In this example we will set the bootstrap to 60.
Open Kallisto-0.42.3-Qaunt-PE (Apps > Operation > Analysis > Gene expression analysis > Kallisto-v.0.43.1)
- Name your analysis.( Give a name of your preference or keep the default)

- Select "Input"
enter a transcript fasta file
Community Data -> iplantcollaborative -> example_data -> Kallisto_Sleuth-> Sorghum_bicolor.Sorbi1.20.cdna.all.fa
input fastq files and select paired option
Community Data -> iplantcollaborative -> example_data -> Kallisto_Sleuth-> reads

- Set the number of bootstraps too 60.

- Click on "Launch Analysis."
- Once the analysis is completed , click on "Analysis," and then click on the analysis "Name" to open the output folder.
Output:

After quantification, you will get a number of files in the output directory for each sample.run_info.json- some high-level information about the run, including the command and versions of kallisto used to generate the outputabundance.tsv- a plain text file with transcript level abundance estimates. This file can be read into R or any other statistical language easily (e.g.read.table('abundance.tsv'))abundance.h5- a HDF5 file containing all of the quantification information including bootstraps and other auxiliary information from the run. This file is read by sleuth
2. Sleuth for analyzing gene level differential expression (once per each experimental sample) and visualization in Atmosphere-
sleuth is a tool for the analysis and comparison of multiple related RNA-Seq experiments. Key features include:
- The ability to perform both transcript-level and gene-level analysis.
- Compatibility with kallisto enabling a fast and accurate workflow from reads to results.
- The use of boostraps to ascertain and correct for technical variation in experiments.
- An interactive app for exploratory data analysis.
Sleuth Paper: Harold Pimentel, Nicolas L Bray, Suzette Puente, Páll Melsted and Lior Pachter, Differential analysis of RNA-seq incorporating quantification uncertainty, in press.
We will use Atmosphere cloud service for doing gene level differential expression and visualization of the above analyzed Kallisto data. We will use the output from the Kallisto step that created the transcript level abundance files.
Community Data -> iplantcollaborative -> example_data -> Kallisto_Sleuth-> kallisto_qaunt_output
Here we are doing a pairwise comparison for differential expression in sensitive genotype(IS20351) under Drought Stress(DS) and Well Watered(WW) condition. We have 3 replicates under each condition
Section 1: Connect to an instance of an Atmosphere Image and launch a instance
1. Go to https://atmo.cyverse.org/ and log in with IPLANT TEST USER CREDENTIALS.

2. Click on images. Search "Ubuntu 16.04 GUI XFCE Base" on the search space. Click on "Ubuntu 16.04 GUI XFCE Base" image.

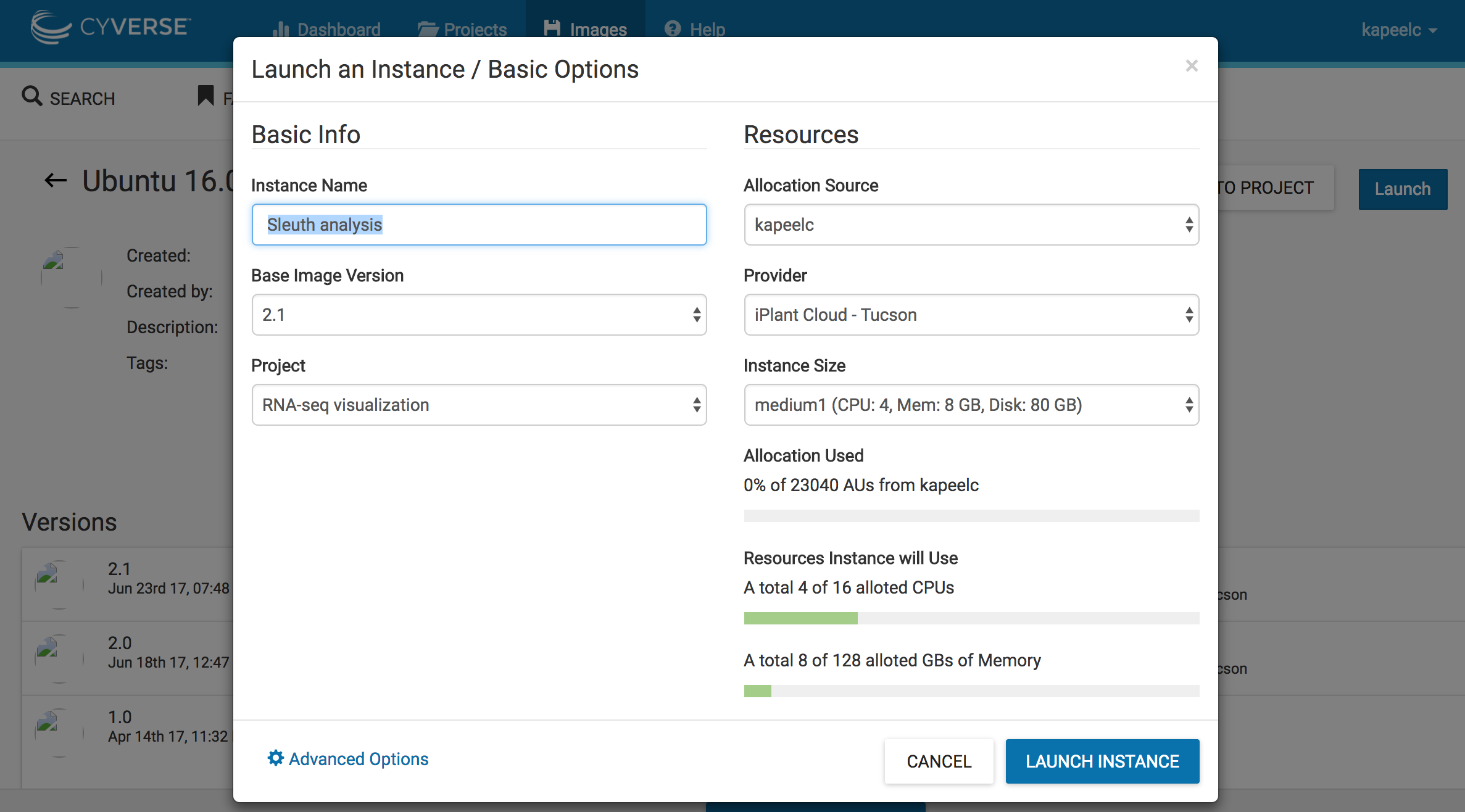
3. Click on Launch. Add to existing projects if you have already created one. In this case, we will add the instance to existing project "RNA-seq visualization". Select the size of the instance "medium 1(CPU:4,Mem: 8, Disk: 60Gb)". Under basic info - > instance name write "Sleuth analysis". Click on Launch instance. This will launch a instance of the image.
4. Once the instance is active, you will get a email confirmation and can also see the status active on the Atmosphere portal

5. Copy the IP address "128.196.64.125", if you are using MAC or Ubuntu open a terminal window and paste the IP address on the command line using your Cyverse user name and hit enter. This will ask you your Cyverse user account passowrd, hit enter
Note- After entering the password if its ask you to add your IP address to the config, enter yes.
escudilla:~ kchougul$ ssh kapeelc@128.196.64.125
After you enter the password you should see something like this
escudilla:~ kchougul$ ssh kapeelc@128.196.64.125
kapeelc@128.196.64.168's password:
Welcome to Ubuntu 16.04.2 LTS (GNU/Linux 4.4.0-72-generic x86_64)
Get cloud support with Ubuntu Advantage Cloud Guest:
http://www.ubuntu.com/business/services/cloud
45 packages can be updated.
0 updates are security updates.
*** System restart required ***
Welcome to
_ _ _
/ \ | |_ _ __ ___ ___ ___ _ __ | |__ ___ _ __ ___
/ _ \| __| '_ ` _ \ / _ \/ __| '_ \| '_ \ / _ \ '__/ _ \
/ ___ \ |_| | | | | | (_) \__ \ |_) | | | | __/ | | __/
/_/ \_\__|_| |_| |_|\___/|___/ .__/|_| |_|\___|_| \___|
|_|
Last login: Mon Jun 19 14:16:47 2017 from 143.48.50.204
**************************************************************************
# A new feature in cloud-init identified possible datasources for #
# this system as: #
# ['OpenStack', 'None'] #
# However, the datasource used was: CloudStack #
# #
# In the future, cloud-init will only attempt to use datasources that #
# are identified or specifically configured. #
# For more information see #
# https://bugs.launchpad.net/bugs/1669675 #
# #
# If you are seeing this message, please file a bug against #
# cloud-init at #
# https://bugs.launchpad.net/cloud-init/+filebug?field.tags=dsid #
# Make sure to include the cloud provider your instance is #
# running on. #
# #
# After you have filed a bug, you can disable this warning by launching #
# your instance with the cloud-config below, or putting that content #
# into /etc/cloud/cloud.cfg.d/99-warnings.cfg #
# #
# #cloud-config #
# warnings: #
# dsid_missing_source: off #
**************************************************************************
Disable the warnings above by:
touch /home/kapeelc/.cloud-warnings.skipy
or
touch /var/lib/cloud/instance/warnings/.skip
kapeelc@vm64-168:~$
6. Now your instance is ready to use. We first download the data needed to run Sleuth. We create a directory sleuth_analysis to download the input files. We will use Rstudio on atmosphere to do the interactive analysis using Sleuth R package. To download the data we will use command line icommands that are installed in this instance for you. Type the below command on the terminal where the instance is opened and enter your cyverse password to begin download
1. kapeelc@vm64-135:~$ mkdir sleuth_analysis; cd sleuth_analysis 2. kapeelc@vm64-168:~$ iget -PVr /iplant/home/shared/iplantcollaborative/example_data/Kallisto_Sleuth/kallisto_qaunt_output . NOTICE: created irodsHome=/iplant/home/kapeelc NOTICE: created irodsCwd=/iplant/home/kapeelc NOTICE: created irodsHome=/iplant/home/kapeelc NOTICE: created irodsCwd=/iplant/home/kapeelc NOTICE: created irodsHome=/iplant/home/kapeelc NOTICE: created irodsCwd=/iplant/home/kapeelc NOTICE: created irodsHome=/iplant/home/kapeelc NOTICE: created irodsCwd=/iplant/home/kapeelc NOTICE: created irodsHome=/iplant/home/kapeelc NOTICE: created irodsCwd=/iplant/home/kapeelc NOTICE: created irodsHome=/iplant/home/kapeelc NOTICE: created irodsCwd=/iplant/home/kapeelc NOTICE: created irodsHome=/iplant/home/kapeelc NOTICE: created irodsCwd=/iplant/home/kapeelc Enter your current iRODS password: 3. kapeelc@vm64-135:~$ iget -PV /iplant/home/shared/iplantcollaborative/example_data/Kallisto_Sleuth/design_matrix . 4. iget -PV /iplant/home/shared/iplantcollaborative/example_data/Kallisto_Sleuth/t2g.txt .
Once the down load is complete you should see the directory which contains the abundance.tsv count files for each sample that will be used in Sleuth for differential expression and plotting
kapeelc@vm64-125:~/sleuth_analysis$ ls -lh total 8.0K -rw-r--r-- 1 kapeelc iplant-everyone 138 Jul 10 20:45 design_matrix drwxr-x--- 8 kapeelc iplant-everyone 4.0K Jul 10 20:44 kallisto_qaunt_output
Section 2: Using R studio on atmosphere to do the interactive analysis using Sleuth R package
- On the command line type below three commands to start a Rstudio and enter your Cyverse password. This will install all the dependencies needed to start Rstudio.
1. kapeelc@vm64-135:~$ sudo apt-get update && sudo apt-get -y install gdebi-core r-base libxml2-dev [sudo] password for kapeelc: 2. kapeelc@vm64-135:~$ wget https://download2.rstudio.org/rstudio-server-1.0.136-amd64.deb 3. kapeelc@vm64-135:~$ sudo gdebi -n rstudio-server-1.0.136-amd64.deb
2. If you want to find what the IP address of the R studio then run below command,
kapeelc@vm64-135:~$ echo My RStudio Web server is running at: http://$(hostname):8787/ My RStudio Web server is running at: http://vm64-125.iplantcollaborative.org:8787/
just copy and paste http://vm64-135.iplantcollaborative.org:8787/ on to the web-browser
3. Enter your Cyverse user name and password to launch the R studio on browser

4. paste the below R commands to begin the analysis. Start File -> New File -> R script, then paste the commands
